STRING API
STRING has an application programming interface (API) which enables you to get the data without using the graphical user interface of the web page. The API is convenient if you need to programmatically access some information but still do not want to download the entire dataset. There are several scenarios when it is practical to use it. For example, you might need to access some interaction from your own scripts or want to incorporate STRING network in your web page.
We currently provide an implementation using HTTP, where the database information is accessed by HTTP requests. Due to implementation reasons, similarly to the web site, some API methods will allow only a limited number of proteins in each query. If you need access to the bulk data, you can download the entire dataset from the download page
There are several methods available through STRING API:
| Method | API method URL | Description |
|---|---|---|
| Mapping identifiers | /api/tsv/get_string_ids? | Maps common protein names, synonyms and UniProt identifiers into STRING identifiers |
| Getting the network image | /api/image/network? | Retrieves the network image with your input protein(s) highlighted in color |
| Retrieving the interaction network | /api/tsv/network? | Retrieves the network interactions for your input protein(s) in various text based formats |
| Getting the interaction partners | /api/tsv/interaction_partners? | Gets all the STRING interaction partners of your proteins |
| Performing functional enrichment | /api/tsv/enrichment? | Gets the results of the Gene Ontology, KEGG pathways, Pfam and InterPro enrichment analysis of your proteins |
| Performing interaction enrichment | /api/tsv/ppi_enrichment? | Tests if your network has more interactions than expected |
Getting started Top ↑
As STRING API works like normal HTTP request you can access it like any other webpage. Just copy/paste the following URL into your browser to get the PNG image of Patched 1 gene network.
https://string-db.org/api/image/network?identifiers=PTCH1
However most likely you will be accessing the API from your scripts or website. Examples of python scripts for each of the API calls are attached at the end of each section.
In order to query with more than one identifier in one call just separate each identifier by new line character "%0d" or "%0a" (without the quotes, case insensitive) like this:
https://string-db.org/api/image/network?identifiers=PTCH1%0dSHH%0dGLI1%0dSMO%0dGLI3
...but before you get started:
- Please be considerate and wait one second between each call, so that our server won't get overloaded.
- Although STRING understands a variety of identifiers and does its best to disambiguate your input it's always better to map them first (see: mapping).
- If known, please always specify from which species your proteins come from. If you do, each of your queries will be answered faster :)
- When calling our API from your website or tools please identify yourself using the caller_identity parameter.
- STRING understands both GET and POST requests. GET requests, although simpler to use, have a character limit, therefore it is recommended to use POST whenever possible.
Mapping identifiers Top ↑
You can call our STRING API with common gene names, various synonyms or even UniProt identifiers and accession numbers. However, STRING may not always understand them which may lead to errors or inconsistencies. Before using other API methods it is always advantageous to map your identifiers to the ones STRING uses. In addition, STRING will resolve its own identifiers faster, therefore your tool/website will see a speed benefit if you use them. For each input protein STRING places the best matching identifier in the first row, so the first line will usually be the correct one.
Call:
https://string-db.org/api/[output-format]/get_string_ids?identifiers=[your_identifiers]&[optional_parameters]
Avaliable output formats:
| Format | Description |
|---|---|
| tsv | tab separated values, with a header line |
| tsv-no-header | tab separated values, without header line |
| json | JSON format |
| xml | XML format |
Avaliable parameters:
| Parameter | Description |
|---|---|
| identifiers | required parameter for multiple items, e.g. DRD1_HUMAN%0dDRD2_HUMAN |
| echo_query | insert column with your input identifier (takes values '0' or '1', default is '0') |
| limit | limits the number of matches per query identifier (best matches come first) |
| species | NCBI taxon identifiers (e.g. Human is 9606, see: STRING organisms). |
| caller_identity | your identifier for us. |
Output fields:
| Field | Description |
|---|---|
| queryItem | (OPTIONAL) your input protein |
| queryIndex | position of the protein in your input (starting from position 0) |
| stringId | STRING identifier |
| ncbiTaxonId | NCBI taxon identifier |
| taxonName | species name |
| preferredName | common protein name |
| annotation | protein annotation |
Example call (resolving "p53" and "cdk2" in human):
https://string-db.org/api/tsv/get_string_ids?identifiers=p53%0dcdk2&species=9606
Example python 2.7 code:
#!/usr/bin/env python
##########################################################
## For a given list of proteins the script resolves them
## (if possible) to the best matching STRING identifier
## and prints out the mapping on screen in the TSV format
###########################################################
import sys
import urllib2
string_api_url = "https://string-db.org/api"
output_format = "tsv-no-header"
method = "get_string_ids"
my_genes = ["p53", "BRACA", "cdk2", "Q99835"] # your protein list
species = "9606"
limit = 1
echo_query = "1"
my_app = "www.awesome_app.org"
## Construct the request
request_url = string_api_url + "/" + output_format + "/" + method + "?"
request_url += "identifiers=" + "%0d".join(my_genes)
request_url += "&" + "species=" + species
request_url += "&" + "limit=" + str(limit)
request_url += "&" + "echo_query=" + echo_query
request_url += "&" + "caller_identity=" + my_app
## Call STRING
try:
response = urllib2.urlopen(request_url)
except urllib2.HTTPError as err:
error_message = err.read()
print error_message
sys.exit()
## Read and parse the results
line = response.readline()
while line:
l = line.split("\t")
input_identifier, string_identifier = l[0], l[2]
print input_identifier + "\t" + string_identifier
line = response.readline()
Getting STRING network image Top ↑
With our API you can retrieve an image of a STRING network of a neighborhood surrounding one or more proteins or ask STRING to show only the network of interactions between your input proteins. All the network flavors (confidence/evidence/action) are accessible through the API. The API can output the image as a PNG (low and high resolution with alpha-channel) or as an SVG (vector graphics that can be modified through scripts or in an appropriate software).
Call:
https://string-db.org/api/[output-format]/network?identifiers=[your_identifiers]&[optional_parameters]
Avaliable output formats:
| Format | Description |
|---|---|
| image | network PNG image with alpha-channel |
| highres_image | high resolution network PNG image with alpha-channel |
| svg | vector graphic format (SVG) |
Avaliable parameters:
| Parameter | Description |
|---|---|
| identifiers | required parameter for multiple items, e.g. DRD1_HUMAN%0dDRD2_HUMAN |
| species | NCBI taxon identifiers (e.g. Human is 9606, see: STRING organisms). |
| add_color_nodes | adds color nodes based on scores to the input proteins |
| add_white_nodes | adds white nodes based on scores to the input proteins (added after color nodes) |
| required_score | threshold of significance to include an interaction, a number between 0 and 1000 (default depends on the network) |
| network_flavor | the style of edges in the network: evidence, confidence (default), actions |
| caller_identity | your identifier for us. |
If you query the API with one protein the "add_white_nodes" parameter is automatically set to 10, so you can see the interaction neighborhood of your query protein. However, similarly to the STRING webpage, whenever you query the API with more than one protein we show only the interactions between your input proteins. You can, of course, always extend the interaction neighborhood by setting "add_color/white_nodes" parameter to the desired value.
Output (EGFR and TP53 actions neighborhood):
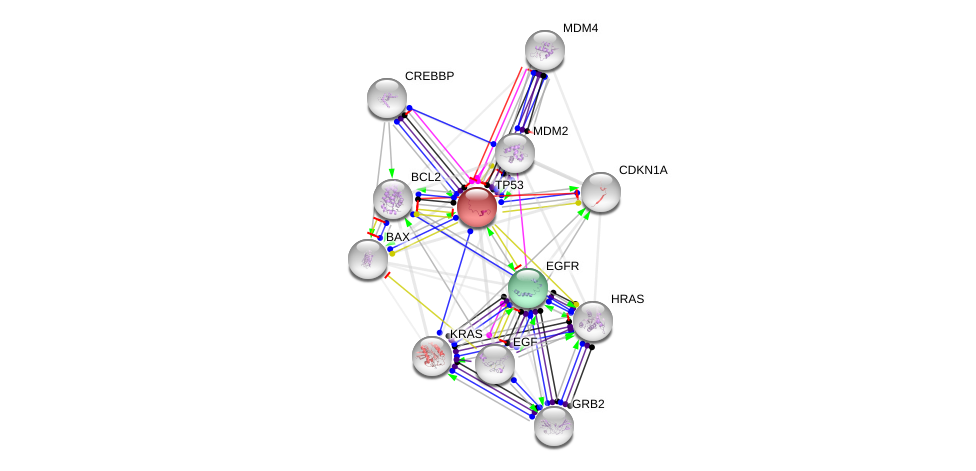
Example call (EGFR and TP53 interaction neighborhood):
https://string-db.org/api/image/network?identifiers=TP53%0dEGFR&add_white_nodes=10&network_flavor=actions
Example python 2.7 code:
#!/usr/bin/env python
###############################################################################
## For each pairs of proteins in a given list save the PNG image of
## STRING network of their 15 most confident interaction partners.
## - make my input protein pairs in color and the niegborhood proteins in white
## - set the network flavour to "confidence"
###############################################################################
import urllib
from time import sleep
string_api_url = "https://string-db.org/api"
output_format = "image"
method = "network"
my_genes = [["YMR055C", "YFR028C"],
["YNL161W", "YOR373W"],
["YFL009W", "YBR202W"]]
species = "4932"
my_app = "www.awesome_app.org"
## Construct the request
request_url = string_api_url + "/" + output_format + "/" + method + "?"
request_url += "identifiers=%s"
request_url += "&" + "species=" + species
request_url += "&" + "add_white_nodes=15"
request_url += "&" + "caller_identity=" + my_app
## For each gene call STRING
for gene_pair in my_genes:
gene1, gene2 = gene_pair
urllib.urlretrieve(request_url % "%0d".join(gene_pair), "%s.png" % "_".join(gene_pair))
sleep(1)
Getting the STRING network interactions Top ↑
The network API method also allows you to retrieve your STRING interaction network for one or multiple proteins in various text formats. It will tell you the combined score and all the channel specific scores for the set of proteins. You can also extend the network neighborhood by setting "add_nodes", which will add, to your network, new interaction partners in order of their confidence.
Call:
https://string-db.org/api/[output-format]/network?identifiers=[your_identifiers]&[optional_parameters]
Avaliable output formats:
| Format | Description |
|---|---|
| tsv | tab separated values, with a header line |
| tsv-no-header | tab separated values, without header line |
| json | JSON format |
| xml | XML format |
| psi-mi | PSI-MI XML format |
| psi-mi-tab | PSI-MITAB format |
Avaliable parameters:
| Parameter | Description |
|---|---|
| identifiers | required parameter for multiple items, e.g. DRD1_HUMAN%0dDRD2_HUMAN |
| species | NCBI taxon identifiers (e.g. Human is 9606, see: STRING organisms). |
| required_score | threshold of significance to include a interaction, a number between 0 and 1000 (default depends on the network) |
| add_nodes | adds a number of proteins with to the network based on their confidence score |
| caller_identity | your identifier for us. |
If you query the API with one protein the "add_nodes" parameter is automatically set to 10, so you can can get the interaction neighborhood of your query protein. However, similarly to the STRING webpage, whenever you query the API with more than one protein the method will output only the interactions between your input proteins. You can, of course, always extend the interaction neighborhood by setting "add_nodes" parameter to the desired value.
Output fields (TSV and JSON formats):
| Field | Description |
|---|---|
| stringId_A | STRING identifier (protein A) |
| stringId_B | STRING identifier (protein B) |
| preferredName_A | common protein name (protein A) |
| preferredName_B | common protein name (protein B) |
| ncbiTaxonId | NCBI taxon identifier |
| score | combined score |
| nscore | gene neighborhood score |
| fscore | gene fusion score |
| pscore | phylogenetic profile score |
| ascore | coexpression score |
| escore | experimental score |
| dscore | database score |
| tscore | textmining score |
To see how the combined score is computed from the partial scores see FAQ
Example call (retrieve all interactions between TP53 EGFR and CDK2):
https://string-db.org/api/tsv/network?identifiers=TP53%0dEGFR%0dCDK2&required_score=400
Example python 2.7 code:
#!/usr/bin/env python
################################################################
## For a given list of proteins single out only the interactions
## which have experimental evidences and print out their score
################################################################
import sys
import urllib2
string_api_url = "https://string-db.org/api"
output_format = "tsv-no-header"
method = "network"
my_genes = ["CDC42","CDK1","KIF23","PLK1",
"RAC2","RACGAP1","RHOA","RHOB"]
species = "9606"
my_app = "www.awesome_app.org"
## Construct the request
request_url = string_api_url + "/" + output_format + "/" + method + "?"
request_url += "identifiers=%s" % "%0d".join(my_genes)
request_url += "&" + "species=" + species
request_url += "&" + "caller_identity=" + my_app
try:
response = urllib2.urlopen(request_url)
except urllib2.HTTPError as err:
error_message = err.read()
print error_message
sys.exit()
## Read and parse the results
line = response.readline()
while line:
l = line.strip().split("\t")
p1, p2 = l[2], l[3]
experimental_score = float(l[10])
if experimental_score != 0:
print "\t".join([p1,p2, "experimentally confirmed (prob. %.3f)" % experimental_score])
line = response.readline()
Getting all the STRING interaction partners of the protein set Top ↑
Diffrently from the network API method, which retrieves only the interactions between the set of input proteins and between their closest interaction neighborhood (if add_nodes parameters is specified), interaction_partners API method provides the interactions between your set of proteins and all the other STRING proteins. The output is avaliable in various text based formats. As STRING network usually has a lot of low scoring interactions, you may want to limit the number of retrieved interaction per protein using "limit" parameter (of course the high scoring interactions will come first).
Call:
https://string-db.org/api/[output-format]/interaction_partners?identifiers=[your_identifiers]&[optional_parameters]
Avaliable output formats:
| Format | Description |
|---|---|
| tsv | tab separated values, with a header line |
| tsv-no-header | tab separated values, without header line |
| json | JSON format |
| xml | XML format |
| psi-mi | PSI-MI XML format |
| psi-mi-tab | PSI-MITAB format |
Avaliable parameters:
| Parameter | Description |
|---|---|
| identifiers | required parameter for multiple items, e.g. DRD1_HUMAN%0dDRD2_HUMAN |
| species | NCBI taxon identifiers (e.g. Human is 9606, see: STRING organisms). |
| limit | limits the number of interaction partners retrieved per protein (most confident interactions come first) |
| required_score | threshold of significance to include a interaction, a number between 0 and 1000 (default depends on the network) |
| caller_identity | your identifier for us. |
Output fields (TSV and JSON formats):
| Field | Description |
|---|---|
| stringId_A | STRING identifier (protein A) |
| stringId_B | STRING identifier (protein B) |
| preferredName_A | common protein name (protein A) |
| preferredName_B | common protein name (protein B) |
| ncbiTaxonId | NCBI taxon identifier |
| score | combined score |
| nscore | gene neighborhood score |
| fscore | gene fusion score |
| pscore | phylogenetic profile score |
| ascore | coexpression score |
| escore | experimental score |
| dscore | database score |
| tscore | textmining score |
To see how the combined score is computed from the partial scores see FAQ
Example call (retrieve best 10 STRING interactions for TP53 and CDK2):
https://string-db.org/api/tsv/interaction_partners?identifiers=TP53%0dCDK2&limit=10
Example python 2.7 code:
#!/usr/bin/env python
#################################################################
## For each protein in a given list print the names of the best 5
## interaction partners.
#################################################################
import sys
import urllib2
string_api_url = "https://string-db.org/api"
output_format = "tsv-no-header"
method = "interaction_partners"
my_genes = ["9606.ENSP00000000233", "9606.ENSP00000000412",
"9606.ENSP00000000442", "9606.ENSP00000001008"]
species = "9606"
limit = 5
my_app = "www.awesome_app.org"
## Construct the request
request_url = string_api_url + "/" + output_format + "/" + method + "?"
request_url += "identifiers=%s" % "%0d".join(my_genes)
request_url += "&" + "species=" + species
request_url += "&" + "limit=" + str(limit)
request_url += "&" + "caller_identity=" + my_app
try:
response = urllib2.urlopen(request_url)
except urllib2.HTTPError as err:
error_message = err.read()
print error_message
sys.exit()
## Read and parse the results
line = response.readline()
while line:
l = line.strip().split("\t")
query_ensp = l[0]
query_name = l[2]
partner_ensp = l[1]
partner_name = l[3]
combined_score = l[5]
print "\t".join([query_ensp, query_name, partner_name, combined_score])
line = response.readline()
Getting functional enrichment Top ↑
STRING maps several databases onto its proteins, this includes: Gene Ontology, KEGG pathways, Pfam domains and InterPro domains. STRING enrichment API method allows you to retrieve the functional enrichment for any set of your input proteins. It will tell you which of your input proteins have an enriched term and the term's description. The API provides the raw p-values, as well as, False Discovery Rate and Bonferroni corrected p-values. The detailed description of the enrichment algorithm can be found here
Call:
https://string-db.org/api/[output_format]/enrichment?identifiers=[your_identifiers]&[optional_parameters]
Avaliable output formats:
| Format | Description |
|---|---|
| tsv | tab separated values, with a header line |
| tsv-no-header | tab separated values, without header line |
| json | JSON format |
| xml | XML format |
Avaliable parameters:
| Parameter | Description |
|---|---|
| identifiers | required parameter for multiple items, e.g. DRD1_HUMAN%0dDRD2_HUMAN |
| species | NCBI taxon identifiers (e.g. Human is 9606, see: STRING organisms). |
| caller_identity | your identifier for us. |
Output fields:
| Field | Description |
|---|---|
| category | term category (e.g. GO Process, KEGG pathways) |
| term | enriched term (GO term, domain or pathway) |
| number_of_genes | number of genes in your input list with the term assigned |
| ncbiTaxonId | NCBI taxon identifier |
| inputGenes | gene names from your input |
| preferredNames | common protein names (in the same order as your input Genes) |
| p_value | raw p-value |
| fdr | False Discovery Rate |
| bonferroni | bonferroni corrected p-value |
| description | description of the enriched term |
STRING shows only the terms with the raw p-value above 0.1
Example call (the network neighborhood of Epidermal growth factor receptor):
https://string-db.org/api/tsv/enrichment?identifiers=trpA%0dtrpB%0dtrpC%0dtrpE%0dtrpGD
Example python 2.7 code:
#!/usr/bin/env python
#######################################################################
## The script retrieves significantly (FDR < 1%) enriched GO process
## annotations for a given set of proteins and outputs the terms
## on the screen in a TSV format
#######################################################################
import sys
import urllib2
import json
string_api_url = "https://string-db.org/api"
output_format = "json"
method = "enrichment"
my_genes = ['7227.FBpp0074373', '7227.FBpp0077451', '7227.FBpp0077788',
'7227.FBpp0078993', '7227.FBpp0079060', '7227.FBpp0079448']
species = "7227"
my_app = "www.aweseome_app.org"
## Construct the request
request_url = string_api_url + "/" + output_format + "/" + method + "?"
request_url += "identifiers=" + "%0d".join(my_genes)
request_url += "&" + "species=" + species
request_url += "&" + "caller_identity=" + my_app
## Call STRING
try:
response = urllib2.urlopen(request_url)
except urllib2.HTTPError as err:
error_message = err.read()
print error_message
sys.exit()
## Read and parse the results
result = response.read()
if result:
data = json.loads(result)
for row in data:
term = row["term"]
preferred_names = ",".join(row["preferredNames"])
fdr = row["fdr"]
description = row["description"]
if fdr < 0.01:
print "\t".join([term, preferred_names, str(fdr), description])
Getting protein-protein interaction enrichment Top ↑
Even in the absence of annotated proteins (e.g. in novel genomes) STRING can tell you if your subset of proteins is functionally related, that is, if it is enriched in interactions in comparison to the background proteome-wide interaction distribution. The detailed description of the PPI enrichment method can be found here
Call:
https://string-db.org/api/[output_format]/ppi_enrichment?identifiers=[your_identifiers]&[optional_parameters]
Avaliable output formats:
| Format | Description |
|---|---|
| tsv | tab separated values, with a header line |
| tsv-no-header | tab separated values, without header line |
| json | JSON format |
| xml | XML format |
Avaliable parameters:
| Parameter | Description |
|---|---|
| identifiers | required parameter for multiple items, e.g. DRD1_HUMAN%0dDRD2_HUMAN |
| species | NCBI taxon identifiers (e.g. Human is 9606, see: STRING organisms). |
| required_score | threshold of significance to include a interaction, a number between 0 and 1000 (default depends on the network) |
| caller_identity | your identifier for us. |
Output fields:
| Field | Description |
|---|---|
| number_of_nodes | number of proteins in your network |
| number_of_edges | number of edges in your network |
| average_node_degree | mean degree of the node in your network |
| local_clustering_coefficient | average local clustering coefficient |
| expected_number_of_edges | expected number of edges based on the nodes degrees |
| p_value | significance of your network having more interactions than expected |
Example call (the network neighborhood of Epidermal growth factor receptor):
https://string-db.org/api/tsv/ppi_enrichment?identifiers=trpA%0dtrpB%0dtrpC%0dtrpE%0dtrpGD
Example python 2.7 code:
#!/usr/bin/env python
##############################################################
## The script prints out the p-value of STRING protein-protein
## interaction enrichment method for the given set of proteins
##############################################################
import sys
import urllib2
string_api_url = "https://string-db.org/api"
output_format = "tsv-no-header"
method = "ppi_enrichment"
my_genes = ['7227.FBpp0074373', '7227.FBpp0077451', '7227.FBpp0077788',
'7227.FBpp0078993', '7227.FBpp0079060', '7227.FBpp0079448']
species = "7227"
my_app = "www.awesome_app.org"
## Construct the request
request_url = string_api_url + "/" + output_format + "/" + method + "?"
request_url += "identifiers=" + "%0d".join(my_genes)
request_url += "&" + "species=" + species
request_url += "&" + "caller_identity=" + my_app
## Call STRING
try:
response = urllib2.urlopen(request_url)
except urllib2.HTTPError as err:
error_message = err.read()
print error_message
sys.exit()
## Read and parse the results
result = response.readline()
if result:
pvalue = result.strip().split("\t")[5]
print pvalue
Additional information about the API Top ↑
If you need to do a large-scale analysis, please download the full data set. Otherwise you may end up fooding the STRING server with API requests. In particular, try to avoid running scripts in parallel :)
Please contact us if you have any questions regarding the API.
API for retrieving abstracts and actions is temporary undocumented but will return soon in a revised form.